In the realm of pharmaceuticals, there are some concerning statistics according to the Pharmaceutical Research and Manufacturers of America. One in ten medicines worldwide are presumed to be counterfeit. In addition, 95% of internet drug outlets have been found to be out of compliance with federal and state pharmacy laws and practice standards. During one week alone in March of 2020, over 48,000 packages containing counterfeit medicines were seized by Interpol.
In serialization, systems assign a unique serial number linked to information about the product origin, batch number, and expiration date to each saleable unit of each prescription drug product. According to a new report by PMMI Business Intelligence, “Pharmaceutical & Medical Devices | Trends & Opportunities in Packaging Operations,” regulations vary worldwide.
In the United States, the federal Drug Supply Chain Security Act (DSCSA), enacted in 2013, establishes a system to track and trace prescription drugs throughout the U.S. supply chain. The key purposes of DSCSA are to verify the legitimacy of the drug product identifier down to the package level; to make drug recalls more efficient; and, to enhance detection of illegitimate products in the drug supply chain.
The goal of DSCSA is unit-level traceability by 2023, including aggregation, or being able to trace a single unit to the larger bundle, case or pallet that it came from. 
Implementing serialization technology creates challenges beyond compliance at all stages of the packaging process due to the numerous steps involved. 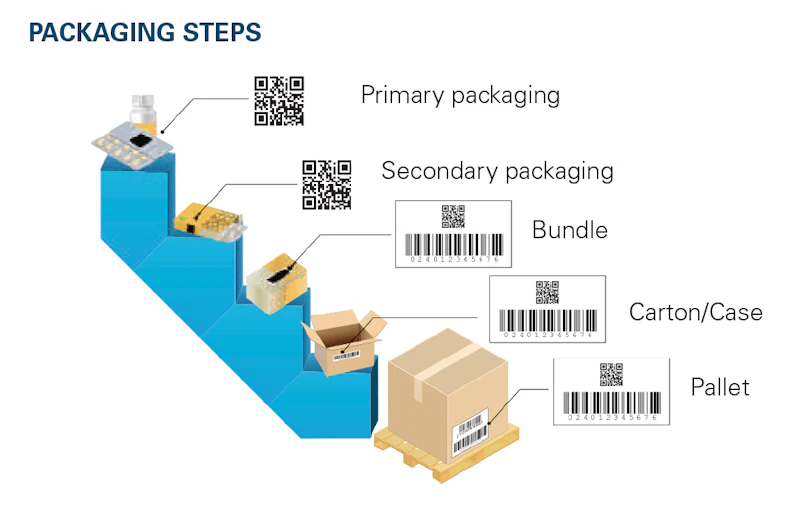
There are specific challenges to serialization in pharmaceutical packaging such as noncompliant or unusable bar codes, the potential for lost productivity on production lines, the need for major human and capital investment for new processes and data management, and inventory issues caused by the mismanaged tracking of returned products.
For medical devices, the FDA established the Unique Device Identification (UDI) system in 2014, to improve patient safety by identifying devices sold in the United States beginning with manufacturing and continuing through distribution and patient use. When fully implemented, the labels on most medical devices will include a UDI in both human-readable, and machine-readable form on the device itself. Device labelers must also submit certain information about each device to the FDA’s Global Unique Device Identification Database (GUDID).
UDI requirements have been implemented in stages, beginning with Class III devices. Due to the impact of COVID-19, the final stage of implementation – for Class I devices – has been postponed from September 2020 to September 2022.
Download a free copy of this white paper below.
Source: PMMI Business Intelligence, “Pharmaceutical & Medical Devices | Trends & Opportunities in Packaging Operations”
To see Pharmaceutical automation, packaging, and materials solutions, check out PACK EXPO Connects online, available through March 31, 2021.
See this 5 minute video: Pharma Track & Trace Lessons Learned
See this 22 minute video: Traceability and Challenges in Perishable Food and E-commerce