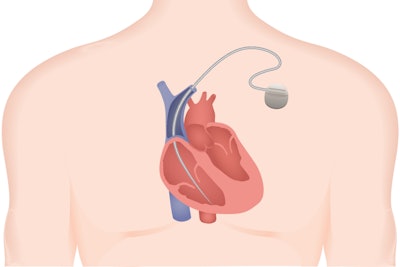
The FDA has a reporting system in order to monitor incidents related to drugs and medical devices to determine if action needs to be taken. According to a recent Ars Technica article, that’s not always the story. Medtronic’s Sprint Fidelis device, which consists of just two wires and a defibrillator, was apparently giving patients random harmful shocks and often failed during cardiac emergencies, and the FDA was hiding the reports from the public.
Malfunction reports are typically recorded in the FDA’s Manufacturer and User Facility Device Experience (MAUDE) database, which is public-facing. However, the FDA quietly set up an alternative reporting log and granted reporting exemptions for a wide range of medical devices. The Sprint Fidelis device was recalled in 2007, after it had been implanted in 268,000 patients, many of whom have since had to make the difficult decision of living with a faulty device, or taking on a life-threatening removal procedure. Since Kaiser Health News broke the story, the FDA announced that it will be shutting down the “alternative summary reporting” repository and ending exemptions.